| Agammaglobulinemia ligada al X | 300755 | BTK | Xq22.1 |
| Albnismo oculocutáneo, tipo II (OCA2) | 203200 | OCA2 | 15q13.1 |
| Alfa Talasemia / Síndrome de retraso mental | 141750 | Multiple | 16p13.3 |
| Alzheimer de inicio temprano con angiopatia amiloide cerebral | 104300 | APP | 21q21.3 |
| Alzheimer de inicio temprano con angipatía amiloide cerebral | 605714 | APP | 21q21.3 |
| Amaurosis congénita de Leber, tipo X (LCA10) | 611755 | CEP290 | 12q21.32 |
| Anemia Diamond-Blackfan 1 | 105650 | RPS19 | 19q13.2 |
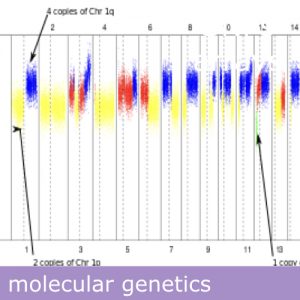
| Aneuploidia completa de cualquier cromosoma | | Multiple | 24 chromosomes |
| Aniridia, tipo II | 106210 | PAX6 | 11p13 |
| Ataxia cerebelar y retraso mental, recesiva (forma Hutterite) | 224050 | VLDLR | 9p24.2 |
| Ataxia de inicio temprano con apraxia oculomotora e hipoalbuminemia, autosómica recesiva | 208920 | APTX | 9p13.3 |
| Autismo con discapacidad intelectual relacionado con SHANK2 | 613436 | SHANK2 | 11q13.3 |
| Baja talla idiopática, ligado al X/Y (ISSX) | 300582 | SHOX | Xp22.33/Yp11.32 (PAR1) |
| Beta talasemia | 613985 | HBB | 11p15.4 |
| Blefarofimosis, ptosis, epicanthus inversus (BPE) | 110100 | FOXL2 | 3q22.3 |
| Cefalopolisindactilia de Greig | 175700 | GLI3 | 7p14.1 |
| Cistinosis nefropática, autosómica recesiva | 219800 | CTNS | 17p13.3 |
| Cohen, autosómica recesiva | 216550 | VPS13B | 8q22.2 |
| Condrodisplasia punctata, recesiva ligada al X (CDPX1) | 302950 | ARSE | Xp22.33 |
| Coroideremia, ligada al X | 303100 | CHM | Xq21.2 |
| Craniosinostosis de Shprintzen-Goldberg | 182212 | FBN1 | 15q21.1 |
| Craniosinostosis, tipo 2 / Craniosinostosis tipo Boston | 604757 | MSX2 | 5q35.2 |
| Craniosinostosis, tipo 2 / Craniosistosis tipo Boston | 604757 | MSX2 | 5q35.2 |
| Defecto del septo atrial (ASD) con defectos en la conducción atrioventricular | 108900 | NKX2-5 | 5q35.2 |
| Deficiencia de esteroide sulfatasa / Ictiosis ligada al X | 308100 | STS | Xp22.31 |
| Deficiencia de Ornitina transcarbamilasa | 311250 | OTC | Xp11.4 |
| Déficit de glicerol quinasa (GKD) | 300679 | GK | Xp21.2 |
| Déficit de GLUT1 | 606777 | SLC2A1 | 1p34.2 |
| Deleción 2q11.2 | | Multiple, LMAN2L, ARID5A | 2q11.2 |
| Deleción 2q11.2-q13 | | Multiple, NCK2, FHL2 | 2q11.2-q13 |
| Deleción Xp11.4-p21.2 | | Multiple, IL1RAPL1, OTC | Xp11.4-p21.2 |
| Discapacidad intelectual con hipoplasia cerebelosa y aspecto facial distinto, ligado al X | 300486 | OPHN1 | Xq12 |
| Discapacidad intelectual con trastornos en el lenguaje y rasgos autistas | 613670 | FOXP1 | 3p14.1 |
| Discapacidad intelectual tipo Siderius, ligada al X | 300263 | PHF8 | Xp11.22 |
| Discapacidad intelectual y microcefalia con pontina e hipoplasia cerebelosa, ligada al X | 300749 | CASK | Xp11.4 |
| Discondrosteosis de Leri-Weill (LWD) | 127300 | SHOX | Xp22.3/Yp11.32 |
| Disgenesia gonadal / Reversión sexual, autosómica dominante (SRA2) | 154230 | Multiple (DMRT1) | 9p24.3 |
| Displasia Campomelica (CMPD) | 114290 | SOX9 | 17q24.3 |
| Displasia Cleidocraneal (CCD) | 119600 | RUNX2 | 6p12.3 |
| Displasia de Kniest / Síndrome de Stickler, tipo I | 156550 | COL2A1 | 12q13.11 |
| Displasia ectodérmica hipohidrotica, ligada al X (XHED) | 305100 | EDA | Xq13.1 |
| Displasia Faciogenital, ligada al X/ Aarskog-Scott, liagado al X | 305400 | FGD1 | Xp11.22 |
| Displasia mesomélica de Langer (LMD) | 249700 | SHOX | Xp22.3/Yp11.32 |
| Displasia mesomélica tipo Kantaputra | 156232 | HOXD gene cluster | 2q31.1 |
| Distonia Dopa-sensible (DRD)/Segawa | 128230 | GCH1 | 14q22.2-q22.3 |
| Distonia mioclonica | 159900 | SGCE | 7q21.3 |
| Distrofia muscular Emery-Dreifuss, liagada al X (EDMD) | 181350 | EMD | Xq28 |
| Dosis anómala de SRY / XX masculino | 278850 | SRY | Yp11.31 |
| Dosis anómala de SRY / XY disgenesia gonadal | 400044 | SRY | Yp11.31 |
| Ectopia lentis aislado | 129600 | FBN1 | 15q21.1 |
| Enfermedad de Charcot-Marie-Tooth, tipo 1A (CMT1A) / Microduplicación 17p12 | 118220 | PMP22 | 17p12 |
| Enfermedad de Norrie, ligada al X | 310600 | NDP | Xp11.3 |
| Enfermedad granulomatosa crónica, ligada al X | 306400 | CYBB | Xp11.4 |
| Enfermedad poliquistica renal 1 (PKD1) | 601313 | PKD1 | 16p13.3 |
| Epilepsia generalizada con convulsiones febriles plus 2 | 604403 | SCN1A | 2q24.3 |
| Epilepsia mioclónica severa de la infancia (SMEI) / Síndome de Dravet | 607208 | SCN1A | 2q24.3 |
| Epilepsia neonatal benigna, | 121200 | KCNQ2 | 20q13.33 |
| Epilepsia y discapacidad intelectual restringido a hembras, ligado al X /Juberg-Hellman, ligado al X / Dravet-like, ligado X | 300088 | PCDH19 | Xq22.1 |
| Esclerosis tuberosa, tipo 1 (TSC1) | 191100 | TSC1 | 9q34.13 |
| Esclerosis tuberosa, tipo 2 (TSC2) | 613254 | TSC2 | 16p13.3 |
| Espasmo infantil, MAGI2-related | | MAGI2 | 7q21.11 |
| Espasmo infantil,CDKL5-related, ligado al X / Rett atípico | 300672 | CDKL5 | Xp22.13 |
| Esquizencefalia | 269160 | EMX2 | 10q26.11 |
| Estatura baja - defectos en el cerebelo e hipófisis - silla turca peque√±a | 262700 | LHX4 | 1q25.2 |
| Fenotipo MASS | 604308 | FBN1 | 15q21.1 |
| Fenotipo Prader-Willi-like | 176270 | Multiple: SIM1 | 6q16.1-q16.3 |
| Foramina parietal, tipo 1 | 168500 | MSX2 | 5q35.2 |
| Haploinsuficiencia ANKRD11 /Microdeleción 16q24.3 | | ANKRD11 | 16q24.3 |
| Haploinsuficiencia EPHA7 / Microdeleción 6q16.1 | | EPHA7 | 6q16.1 |
| Haploinsuficiencia NFIA / Síndrome de microdeleción 1p32p31 | 613735 | NFIA | 1p31.3 |
| Hemidisplasia congénita con eritrodermia ictisiforme y defectos de las extremidades (CHILD) | 308050 | NSDHL | Xq28 |
| Hemofilia A, ligada al X | 306700 | F8 | Xq28 |
| Hemofilia B, ligada al X | 306900 | F9 | Xq27.1 |
| Hernia diafragmática congénita (CDH) | 142340 | Multiple: CHD2, NR2F2 | 15q26.1-q26.3 |
| Heterotaxia, ligada al X | 306955 | ZIC3 | Xq26.3 |
| Hidrocefalia con diabetes insipida nefrogenica, liagada al X | | L1CAM, AVPR2 | Xq28 |
| Hiper-IgE por infección recurrente, autosómica recesiva | 243700 | DOCK8 | 9p24.3 |
| Hipercalcemia hipocalci√∫rica familar, tipo 1(HHC1) | 145980 | CASR | 3q21.1 |
| Hiperplasia adrenal congénita (CAH) | 201910 | CYP21A2 | 6p21.32 |
| Hipoparatiroidismo primario neonatal severo (NSHPT) | 239200 | CASR | 3q21.1 |
| Hipoplasia adrenal congénita (AHC) | 300200 | NR0B1 (DAX1) | Xp21.2 |
| Hipoplasia dérmica focal, ligada al X/Síndrome Gorlin-Goltz | 305600 | PORCN | Xp11.23 |
| Hipospadias 2 aislado, ligado al X | 300758 | MAMLD1 | Xq28 |
| Hipotonia-cistinuria | 606407 | SLC3A1, PREPL | 2p21 |
| Hirschsprung, RET-related | 142623 | RET | 10q11.21 |
| Holoprosencefalia, tipo 1 (HPE1) | 236100 | Multiple | 21q22.3 |
| Holoprosencefalia, tipo 2 (HPE2) | 157170 | SIX3 | 2p21 |
| Holoprosencefalia, tipo 3 (HPE3) | 142945 | SHH | 7q36.3 |
| Holoprosencefalia, tipo 4 (HPE4) | 142946 | TGIF | 18p11.31 |
| Holoprosencefalia, tipo 5 (HPE5) | 609637 | ZIC2 | 13q32.3 |
| Holoprosencefalia, tipo 6 | 605934 | Multiple | 2q37.1-q37.3 |
| Holoprosencefalia, tipo 7 (HPE7) | 610828 | PTCH1 | 9q22.32 |
| Holoprosencefalia, tipo 8 (HPE8) | 609408 | Multiple | 14q13.1-q13.2 |
| insensibilidad a hormona del crecimiento, autosómica recesiva / Laron, autosómica recesiva | | HMGA2 | 12q14.3 |
| Insensibilidad a hormona del crecimiento, autosómica recesiva / Laron, autosómica recesiva | 262500 | GHR | 5p12 |
| Leiomiomatosis, difusa, con Síndrome de Alport (DL-ATS) | 308940 | COL4A5, COL4A6 | Xq22.3 |
| Lesch-Nyhan, ligada al X (LNS) | 300322 | HPRT1 | Xq26.2 |
| Leucodistrofia de aparición adulta autosómica dominante (ADLD) | 169500 | LMNB1 | 5q23.2 |
| Leucodistrofia metacromática, autosómica recesiva (MLD)/Déficit de Arylsulfatase A | 250100 | ARSA | 22q13.3 |
| Síndrome de Li-Fraumeni, tipo 1 (LFS) | 151623 | TP53 | 17p13.1 |
| Lisencefalia con genitales ambiguos,ligado al X 2 (LISX2) | 300215 | ARX | Xp21.3 |
| Lisencefalia con hipoplasia cerebelosa | 257320 | RELN | 7q22.1 |
| Lisencefalia, ligada al X | 300067 | DCX | Xq22.3 |
| Lisencefalia, tipo 1 | 607432 | PAFAH1B1 (LIS1) | 17p13.3 |
| Loeys-Dietz syndrome type 1B (LDS), TGFBR1-related | 610168 | TGFBR2 | 3p24.1 |
| Malformación cavernosa cerebral, tipo 1 (CCM1) | 116860 | KRIT1 | 7q21.2 |
| Malformación cavernosa cerebral, tipo 2 (CCM2) | 603284 | CCM2 | 7p13 |
| Malformación cavernosa cerebral, tipo 3 (CCM3) | 603285 | PDCD10 | 3q26.1 |
| Mano hendida/ malformación pie, tipo 1 (SHFM1) / Ectrodactilia | 183600 | Multiple, SHFM1 | 7q21.3 |
| Mano hendida/ malformación pie, tipo 3 (SHFM3) / Ectrodactilia | 246560 | Multiple, FBXW4 | 10q24.32 |
| Mano hendida/ malformación pie, tipo 4 (SHFM4) | 605289 | TP63 | 3q28 |
| Mano hendida/ malformación pie, tipo 5 (SHFM5) | 606708 | EVX2, HOXD cluster | 2q31.1 |
| Microdelción 5q22 / Poliposis adenomatosa familiar (FAP) | 175100 | Multiple, APC | 5q22.2 |
| Microdeleción 14q32.2 causante de fenotipo upd(14) mat | | Multiple, DLK1, RTL1 | 14q32.2 |
| Microdeleción 14q32.2 causante de fenotipo upd(14)pat | 608149 | Multiple, MEG3, MEG8, RTL1 | 14q32.2 |
| Microdeleción 15q25.2 | | Multiple | 15q25.2 |
| Microdeleción 16p13.11 | | Multiple, MYH11 | 16p13.11 |
| Microdeleción 17q12 | 614527 | Multiple, HNF1B, LHX1 | 17q12 |
| Microdeleción 1q21.1 con susceptibilidad a trombocitopenia y aplasia radial (TAR) | 274000 | Multiple | 1q21.1 |
| Microdeleción 1q41-q42 /Síndrome de Fryns | 612530 | Multiple | 1q41-q42 |
| Microdeleción 2p16.3 /Pitt-Hopkins-like 2 | 614325 | NRXN1 | 2p16.3 |
| Microdeleción 2q23.1 | | MBD5, EPC2 | 2q23.1 |
| Microdeleción 2q37 | 600430 | Multiple | 2q37 |
| Microdeleción 5q14.3 | 613443 | MEF2C | 5q14.3 |
| Microdeleción 5q14.3 | 613443 | MEFC2 | 5q14.3 |
| Microdeleción 6q24.3 | 612863 | Multiple | 6q24.3 |
| Microdeleción 9q34 / Síndrome de Kleefstra | 610253 | EHMT1 | 9q34 |
| Microdeleción distal 16p11.2 | 613444 | Multiple, SH2B1 | 16p11.2 |
| Microdeleción distal 17p13.3 | | YWHAE, CRK | 17p13.3 |
| Microdeleción distal 17p13.3 sin incluir la región de lisencefalia (PAFAH1B1) | 613215 | YWHAE, CRK | 17p13.3 |
| Microdeleción distal 1q21.1 (con susceptibilidad a retraso mental, autismo o anomalías congénitas) | 612474 | Multiple, ACP6, GJA5, GJA8 | 1q21.1 |
| Microdeleción Xp11.3 | 300578 | Multiple, ZNF674, RP2 | Xp11.3 |
| Microduplicación 12q24.21-q24.23 | | Multiple | 12q24.21-q24.23 |
| Microduplicación 15q11-q13 | 608636 | Multiple | 15q11-q13 |
| Microduplicación 16p13.3 | 613458 | CREBBP | 16p13.3 |
| Microduplicación 3q29 | 611936 | Multiple | 3q29 |
| Microduplicación 5p13 | 613174 | NIPBL | 5p13.2 |
| Microduplicación 5q35.2-q35.3 | | NSD1 | q35.2-q35.3 |
| Microduplicación 8p23.1 | | Multiple, GATA4 | 8p23.1 |
| Microduplicación 8q12 | | Multiple, CHD7 | 8q12 |
| Microduplicación distal 17p13.3 incluyendo la región de lisencefalia (PAFAH1B1) | 613215 | PAFAH1B1, YWHAE, CRK | 17p13.3 |
| Microduplicación Xq28 | 300815 | Multiple, GD1, IKBKG | Xq28 |
| Microftalmia 7 con defectos cutáneos lineales (MCOPS7) | 309081 | HCCS | Xp22.2 |
| Microftalmia sindromica, tipo 3 (MCOPS3) | 206900 | SOX2 | 3q26.33 |
| Miopatía miotubular 1, ligada al X | 310400 | MTM1 | Xq28 |
| Nefronoptisis, tipo 1 | 256100 | NPHP1 | 2q13 |
| Neurofibromatosis, tipo 1 (NF1) / Enfermedad de Von Reckinghausen | 162200 | Multiple - NF1 | 17q11.2 |
| Neurofibromatosis, tipo 2 (NF2) | 101000 | NF2 | 22q11.2 |
| Neuropatía hereditaria con predisposición a la parálisis por presión (HNPP)/ Microdeleción 17p12 | 162500 | PMP22 | 17p12 |
| Nevus de células basales/Síndrome de Gorlin-Goltz | 109400 | PTCH1 | 9q22.32 |
| Oculofaciocardiodental, ligado al X / Microftalmia 2 | 300166 | BCOR | Xp11.4 |
| Osteopatia estriada con esclerosis cranial,ligada al X | 300373 | FAM123B | Xq11.1 |
| Paladar hendido, aislado (CPI) | 119540 | SATB2 | 2q33.1 |
| Paraganglioma-feocromocitoma hereditario, SDHB-related | 115310 | SDHB | 1p36.13 |
| Paraganglioma-feocromocitoma hereditario, SDHD-related | 168000 | SDHD | 11q23.1 |
| Pelizaeus-Merzbacher (PMD), ligado al X | 312080 | PLP1 | Xq22.2 |
| Pitt-Hopkins-like 1, autosómica recesiva / Epilepsia Cortical dysplasia-focal epilepsy, autosomal recessive (CDFE syndrome) | 610042 | CNTNAP2 | 7q35-q36.1 |
| Polimicrogiria frontoparietal bilateral (BFPP) | 606854 | GPR56 | 16q13 |
| Poliposis adenomatosa familiar por microdeleción de 5q22 /Síndrome de Gardner | 175100 | APC | 5q22 |
| Quistes renales y diabetes (RCAD) | 137920 | HNF1B | 17q12 |
| Retinoblastoma con discapacidad intelectual | 180200 | RB1 | 13q14.2 |
| Retinoquistis juvenil, ligado al X | 312700 | RS1 | Xp22.13 |
| Retras mental 21, ligado al X | 300143 | IL1RAPL1 | Xp21.3 |
| Retraso mental 1, autosómica dominante (MRD1) | 156200 | MBD5 | 2q23.1 |
| Retraso mental 30, ligado al X | 300558 | PAK3 | Xq22.3 |
| Retraso mental 5, autosómica dominante | 612621 | SYNGAP1 | 6p21.32 |
| Retraso mental 54, ligado al X | 300419 | ARX | Xp21.3 |
| Retraso mental 6, autosómica recesiva | 611092 | GRIK2 | 6q16.3 |
| Retraso mental 7, autosómica recesiva | 611093 | TUSC3 | 8p22 |
| Retraso mental 9, ligado al X/Retraso mental 44, ligado al X | 309549 | FTSJ1 | Xp11.23 |
| Retraso mental 94, ligado al X | 300699 | GRIA3 | Xq25 |
| Retraso mental con déficit aislado de hormona del crecimiento, ligado al X | 300123 | SOX3 | Xq27.1 |
| Retraso mental, ligado al X 17/31, microduplicación | 300705 | Multiple, HSD17B10, HUWE1 | Xp11.22 |
| Rett, variante congénita | 613454 | FOXG1 | 14q12 |
| Reversion sexual sensible a dosis, ligado al X | 300018 | NR0B1 (DAX1) | Xp21.2 |
| Reversión sexual XY 3; (SRXY3 ), +/- insuficiencia adrenal | 612965 | NR5A1 | 9q33.3 |
| Rieger 1 (RIEG1)/Axenfeld-Rieger | 180500 | PITX2 | 4q25 |
| Sindactilia de los dedos de los pies, telecantus, malformaciones anogenitales y renales, ligado al X (STAR) | 300707 | FAM58A | Xq28 |
| Síndorme de Mowat-Wilson | 235730 | ZEB2 | 2q22.3 |
| Síndrome Branquio-oto-renal (BOR)/Síndrome de Melnick-Fraser | 113650 | EYA1 | 8q13.3 |
| Síndrome CardioFacioCutaneo (CFC) | 115150 | Multiple | 7q34, 12p12.1, 19p13.3 |
| Síndrome Craniofrontonasal (CFNS) | 304110 | EFNB1 | Xq13.1 |
| Síndrome Cri-du-Chat | 123450 | Multiple: TERT, CTNND2 | 5p15.2 |
| Síndrome Currarino | 176450 | MNX1 | 7q36.3 |
| Síndrome Dandy-Walker (DWS) | 220200 | Multiple (ZIC1,ZIC4) | 3q24 |
| Síndrome de Alagille, tipo 1 | 118450 | JAG1 | 20p12.2 |
| Síndrome de Alport ligado al X | 301050 | COL4A5 | Xq22.3 |
| Síndrome de Angelman | 105830 | Multiple: UBE3A | 15q11-q13 |
| Síndrome de Bannayan-Riley-Ruvalcaba | 153480 | PTEN | 10q23.31 |
| Síndrome de Barakat / Hipoparatiroidismo, sordera neurosensorial y enfermedad renal (HDR) | 146255 | GATA3 | 10p14 |
| Síndrome de Bardet-Biedl 14 (BBS) | 209900 | CEP290 | 12q21.32 |
| Síndrome de Bartter, tipo 1, antenatal | 146200 | SLC12A1 | 15q21.1 |
| Síndrome de Bartter, tipo 2, antenatal | 241200 | KCNJ1 | 11q24.3 |
| Síndrome de Bartter, tipo 3, clasico | 607364 | CLCNKB | 1p36.13 |
| Síndrome de Bartter, tipo 4 (infanti con sordera neurosensorial) | 602522 | Multiple: CLCNKA, CLCNKB, BSND | 1p32.3 |
| Síndrome de Beckwith-Wiedemann (BWS) | 130650 | KCNQ1OT1, IGF2 | 11p15.5 |
| Síndrome de Beckwith-Wiedemann (BWS), IGF2-related | 130650 | IGF2 | 11p15.5 |
| Síndrome de braquidactilia-retraso mental/ Osteodistrofia hereditaria de Albright | 600430 | HDAC4 | 2q37.3 |
| Síndrome de Buschke-Ollendorff | 166700 | LEMD3 | 12q14.3 |
| Síndrome de Charge | 214800 | CHD7 | 8q12.2 |
| Síndrome de Cornelia de Lange | 122470 | NIPBL | 5p13.2 |
| Síndrome de Cornelia de Lange 2 | 300590 | SMC1A | Xp11.22 |
| Síndrome de Cornelia de Lange, ligada al X | 300590 | SMC1L1 | Xp11.22 |
| Síndrome de Costello | 218040 | HRAS | 11p15.5 |
| Síndrome de Cowden / PTEN tumor hamartoma | 158350 | PTEN | 10q23.31 |
| Síndrome de deleción 10q11.21-q11.23 | | Multiple, CHAT, SLC18A3 | 10q11.21-q11.23 |
| Síndrome de deleción 10q22-q23 | | Multiple | 10q22.3-q23.31 |
| Síndrome de deleción 15q26-qter | 612626 | Multiple | 15q26 |
| Síndrome de deleción 16p13.3 deletion syndrome/Síndrome de Rubinstein-Taybi severo | 610543 | Multiple, CREBBP | 16p13.3 |
| Síndrome de deleción 1p36 | 607872 | Multiple | 1p36 |
| Síndrome de deleción 1q43-q44 | 612337 | Multiple, AKT3 | 1q44 |
| Síndrome de deleción 3q29 | 609425 | Multiple, FBX045, DLG1, PAK2 | 3q29 |
| Síndrome de deleción distal 7q11.23 | 613729 | Multiple | 7q11.23 |
| Síndrome de deleción homocigota 11p15-p14 | 606528 | USHC1, ABCC8 | 11p15.1 |
| Síndrome de DiGeorge / Síndrome VeloCardioFacial / Síndrome deleción 22q11.2 | 188400 | Multiple: HIRA, TBX1 | 22q11.21 |
| Síndrome de DiGeorge, tipo 2 | 601362 | Multiple | 10p14 |
| Síndrome de duplicación 22q11.21 | 608363 | Multiple, TBX1 | 22q11.21 |
| Síndrome de duplicación Xp11.23-p11.22 | 300801 | Multiple | Xp11.23-p11.22 |
| Síndrome de espasmo infantil 1 (ISSX1), ligado al X /Encefalopatía epiléptica infantil temprana 1 | http://omim.org/entry/308350?search=308350&hi | ARX | Xp21.3 |
| Síndrome de Feingold | 164280 | MYCN | 2p24.3 |
| Síndrome de Gitelman | 263800 | SLC12A3 | 16q13 |
| Síndrome de Holt-Oram (HOS) | 142900 | TBX5 | 12q14.21 |
| Síndrome de insensibilidad a los andrógenos | 300068 | AR | Xq12 |
| Síndrome de Jacobsen / Deleción terminal 11q | 147791 | Multiple | 11q23-qter |
| Síndrome de Joubert, tipo 3 | 608629 | AHI1 | 6q23.3 |
| Síndrome de Joubert, tipo 4 (autosómica recesiva) / Síndrome de Joubert, tipo 4 / Síndrome de deleción 2q13 | 609583 | NPHP1 | 2q13 |
| Síndrome de Joubert, tipo 5 (JBTS5) | 610188 | CEP290 | 12q21.32 |
| Síndrome de Kallmann, tipo 1 | 308700 | KAL1 | Xp22.31 |
| Síndrome de Langer-Giedion / Síndrome tricorinofalangeal, tipo II | 150230 | EXT1, TRPS1 | 8q24.11q23.3 |
| Síndrome de Loeys-Dietz tipo 1A (LDS), TGFBR1-related | 609192 | TGFBR1 | 9q22.33 |
| Síndrome de Lowe | 309000 | OCRL | Xq25 |
| Síndrome de Lubs / Síndrome de duplicación MECP2 | 300260 | MECP2 | Xq28 |
| Síndrome de macrocefalia-autismo | 605309 | PTEN | 10q23.31 |
| Síndrome de Marfan, tipo 1 (MFS1) | 154700 | FBN1 | 15q21.1 |
| Síndrome de Marfan, tipo 2 (MFS2) | 610380 | TGFBR2 | 3p24.1 |
| Síndrome de Mc Leod Neuroacantocitosis | 314850 | XK | Xp21.1 |
| Síndrome de Meckel tipo 4 | 611134 | CEP290 | 12q21.32 |
| Síndrome de Menkes | 309400 | ATP7A | Xq21.1 |
| Síndrome de microdeleción 12q14.1-q15 | | Multiple, GRIP1, LEMD3, HMGA2 | 12q14.3 |
| Síndrome de microdeleción 14q11.2 | 613457 | Multiple | 14q11.2 |
| Síndrome de microdeleción 14q22-q23 | | Multiple | 14q22-q23 |
| Síndrome de microdeleción 15q13.3 | 612001 | Multiple, CHRNA7 | 15q13.3 |
| Síndrome de microdeleción 15q24 | 613406 | Multiple | 15q24.1-q24.3 |
| Síndrome de microdeleción 16p11.2 (susceptibilidad ASD) | 611913 | Multiple | 16p11.2 |
| Síndrome de microdeleción 16p11.2-p12.2 | 613604 | Multiple | 16p11.2-p12.2 |
| Síndrome de microdeleción 16p12.1 | 136570 | Multiple, CDR2, EEF2K, UQCRC2 | 16p12.1 |
| Síndrome de microdeleción 16q11.2-q12.2 | | Multiple, SALL1, ZNF423 | 16q11.2-q12.2 |
| Síndrome de microdeleción 16q21-q22 | 614541 | CBFB | 16q21-q22 |
| Síndrome de microdeleción 17p13.1 | 613776 | Multiple, TP53 | 17p13.1 |
| Síndrome de microdeleción 17q21.31 | 610443 | Multiple, MAPT | 17q21.3 |
| Síndrome de microdeleción 17q23.1-q23.2 | 613355 | Multiple, TBX2, TBX4 | 17q23.1-q23.2 |
| Síndrome de microdeleción 19p13.12 | 613638 | Multiple | 19p13.2 |
| Síndrome de microdeleción 19q13.11 | 613026 | Multiple | 19q13.11 |
| Síndrome de microdeleción 22q13.3 (Síndrome de Phelan-McDermid) | 606232 | Multiple, SHANK3 | 22q13.3 |
| Síndrome de microdeleción 2p15-p16.1 | 612513 | Multiple | 2p15-p16.1 |
| Síndrome de microdeleción 2q32.2-q33 | 612313 | Multiple, SATB2 | 2q32.2-q33 |
| Síndrome de microdeleción 6pter-p24 | 612582 | Multiple | 6pter-p24 |
| Síndrome de microdeleción 6q24-q25 | 612863 | Multiple | 6q24-q25 |
| Síndrome de microdeleción 8p23.1 /Hernia diafragmática congénita 2 (CDH2) | 22400 | Multiple, GATA4 | 8p23.1 |
| Síndrome de microdeleción 8q21.11 | 614230 | 8q21.11 | Multiple: ZFHX4 |
| Síndrome de microdeleción 9q22.32-q22.33 | | Multiple, TGFBR1, GABBR2 | 9q22.32-q22.33 |
| Síndrome de microdeleción distal 22q11.2 | 611867 | Multiple | 22q11.21 |
| Síndrome de microdeleción FMR1 | 300624 | FMR1 | Xq27.3 |
| Síndrome de microdeleción homocigota 2p21 | 606407 | Multiple | 2p21 |
| Síndrome de microduplicación 14q12 | | Multiple, FOXG1 | 14q12 |
| Síndrome de microduplicación 15q24 | 613406 | Multiple | 15q24.1-q24.3 |
| Síndrome de microduplicación 17p13.3 | 613215 | Multiple, PAFAH1B1, YWHAE, CRK | 17p13.3 |
| Síndrome de microduplicación 17q12 | 614526 | Multiple, HNF1B, LHX1 | 17q12 |
| Síndrome de microduplicación 17q21.31 | 613533 | Multiple | 17q21.31 |
| Síndrome de microduplicación 7q11.23 | 609757 | Multiple | 7q11.23 |
| Síndrome de Mieller-Diecker | 247200 | PAFAH1B1 (LIS1) | 17p13.3 |
| Síndrome de Mohr-Tranebjaerg | 304700 | TIMM8A | Xq22.1 |
| Síndrome de Nail-patella (NPS) | 161200 | LMX1B | 9q33.3 |
| Síndrome de Noonan, tipo 1 (NS1) | 163950 | PTPN11 | 12q24.13 |
| Síndrome de Noonan, tipo 4 (NS4) | 610733 | SOS1 | 2p22.1 |
| Síndrome de Okihiro / Síndrome de Duane-Radial Ray (DRRS) | 607323 | SALL4 | 20q13.2 |
| Síndrome de Peutz-Jeghers | 175200 | STK11 | 19p13.3 |
| Síndrome de Pitt-Hopkins | 610954 | TCF4 | 18q21.2 |
| Síndrome de poliposis juvenil (JPS), BMPR1A-related | 174900 | BMPR1A | 10q23.2 |
| Síndrome de poliposis juvenil (JPS), SMAD4-related | 174900 | SMAD4 | 18q21.2 |
| Síndrome de Potocki-Lupski (PTLS) | 610883 | Multiple, RAI1 | 17p11.2 |
| Síndrome de Potocki-Shaffer | 601224 | EXT2, ALX4 | 11p11.2 |
| Síndrome de Prader-Willi | 176270 | Multiple | 15q11-q13 |
| Síndrome de Proteus/Proteus-like | 176920 | PTEN | 10q23.31 |
| Síndrome de Rett | 312750 | MECP2 | Xq28 |
| Síndrome de Saethre-Chotzen | 101400 | TWIST1 | 7p21.1 |
| Síndrome de Senior-Loken, tipo 6 (SLSN6) | 610189 | CEP290 | 12q21.32 |
| Síndrome de sobrecrecimiento 15q26 | | Multiple | 15q26 |
| Síndrome de Sotos | 117550 | NSD1 | 5q35.3 |
| Síndrome de Stickler, tipo I | 108300 | COL2A1 | 12q13.11 |
| Síndrome de Townes-Brocks (TBS) | 107480 | SALL1 | 16q12.1 |
| Síndrome de Ulnar-Mammary (UMS) | 181450 | TBX3 | 12q24.21 |
| Síndrome de Van der Woude | 119300 | IRF6 | 1q32.2 |
| Síndrome de Von Hippel-Lindau | 193300 | VHL | 3p25.3 |
| Síndrome de Waardenburg, tipo I | 193500 | PAX3 | 2q36.1 |
| Síndrome de Waardenburg, tipo IIA | 193510 | MITF | 3p14.1 |
| Síndrome de WAGR (tumor de Wilms+Aniridia+Anomalias genitourinarias+Retraso Mental) | 194072 | PAX6, WT1 | 11p13 |
| Síndrome de Walker-Warburg (WWS) | 236670 | LARGE | 22q12.3 |
| Síndrome de Weill-Marchesani 2 | 608328 | FBN1 | 15q21.1 |
| Síndrome de Williams-Beuren | 194050 | Multiple, ELN | 7q11.23 |
| Síndrome de Wolf-Hirschhorn (WHS) | 194190 | Multiple | 4p16.3 |
| Síndrome facila tipo "máscara de Nablus" | 608156 | Multiple | 8q22.1 |
| Síndrome FG, tipo 5 (FGS5) | 300581 | MID2 | Xq22.3 |
| Síndrome hamartoma PTEN | | PTEN | 10q23.31 |
| Síndrome linfoproliferativo, ligado al X (XLP) | 308240 | SH2D1A | Xq25 |
| Síndrome ojo de gato (Cat-Eye) | 115470 | Multiple | 22q11.1 |
| Síndrome Opitz GBBB, ligado al X | 300000 | MID1 | Xp22.2 |
| Síndrome Oral-facial-digital tipo 1 | | OFD1 | Xp22.2 |
| Sindrome Oto-dental | 166750 | FGF3 | 11q13.3 |
| Síndrome Oto-facio-cervical (OFC) | 166780 | EYA1 | 8q13.3 |
| Síndrome Pallister-Killian / Tetrasomia 12p | 601803 | Multiple | 12p |
| Síndrome Rubinstein-Taybi (RTS) | 180849 | CREBBP | 16p13.3 |
| Síndrome Rubinstein-Taybi 2 | 613684 | EP300 | 22q13.2 |
| Síndrome Simpson-Golabi-Behmel, tipo 1 | 312870 | GPC3 | Xq26.2 |
| Síndrome Smith-Lemli-Opitz (SLOS) | 270400 | DHCR7 | 11q13.4 |
| Síndrome Smith-Magenis | 182290 | Multiple, RAI1 | 17p11.2 |
| Síndrome Tricorinofalangeal, tipo I | 190350 | TRSP1 | 8q23.3 |
| Sinpolidactilia / Sindactilia II | 186000 | HOXD cluster | 2q31.1 |
| Sordera 22, autosómica recesiva | 607039 | OTOA | 16p12.2 |
| Sordera neurosensorial, autosómica recesiva | 220290 | GJB6 | 13q12.11 |
| Telangectasia hemorrágica hereditaria, tipo 2 (HHT2) | 600376 | ACVRL1 | 12q13.13 |
| Todas las regiones eucromáticas-pericentroméricas | | Multiple | 43 sites |
| Todas las regiones subteloméricas | | Multiple | 41 sites |
| Trastorno del habla/lenguaje, tipo 1 | 602081 | FOXP2 | 7q31.1 |
| Trastornos asociados al factor de crecimiento vascular endotelial (VEGFA) | 192240 | VEGFA | 6p21.1 |
| Tumor de Wilms, tipo 1 | 194070 | WT1 | 11p13 |
| Usher IIC, autosómica recesiva | 605472 | GPR98 | 5q14.3 |